
今回は、実際にGROMACSで分子動力学計算を行っていきます。前回の記事「GROMACS計算の下準備 ~分子構造作成・力場割り当て・分子の集合体の準備~」で作成したインプットファイルを使いますので、作成していない場合はそちらの記事を参照してください。また、本来は長時間の計算を行う必要がありますが、ここでは流れをまず理解していただくために、短めの計算としておりますことをご了承ください。計算をどのくらい実施する必要があるのかについては、別途記事を書いていきます。
計算の目的と流れについて
改めて、計算の目的と流れについておさらいしましょう。前回の記事「GROMACS計算の下準備 ~分子構造作成・力場割り当て・分子の集合体の準備~」で、『液体のベンゼンの密度を計算によって求める』という課題を設定しました。この液体のベンゼンの密度を求めるには、液体状態でかつ平衡状態となっているベンゼンの集合体を計算する必要があります。
計算の流れについては、過去記事「分子動力学法の計算の流れ」でまとめた通りになります。緩和計算→平衡化計算→サンプリング計算、この順序で進めていきます。平衡化計算が完了した時点で平衡状態のベンゼンの集合体が得られており、そこから一定時間のサンプリング計算を行います。この流れに沿って、計算の仕方を説明していきます。
緩和計算
最初に分子の集合体を安定な状態に持っていく計算を行います、これを一般的に緩和計算と呼びます。packmolで作った集合体構造は安定な構造とはかけ離れているので、ある程度安定な構造にしてから平衡化計算に進めていきます。
緩和計算は3段階に分けて進めていきます。エネルギー極小化計算、NVT計算、NPT計算の順になります。それぞれの計算の解説については、別途記事を作成します。まずはエネルギー極小化計算のインプットを作りましょう。テキストファイルを「minimize.mdp」の名前で作成し、以下の内容をコピペしてください。
;##### Run Control #############
integrator=steep
nsteps=-1
;##### Neighbor searching ######
rlist=1.0
;##### Van der Waals ###########
rvdw=1.0
;##### Electrostatics ##########
rcoulomb=1.0
;##### Energy minimization #####
emtol=10.0
emstep=0.01
;##### Output control ##########
energygrps=System作成したら、以下のコマンドを実行してください、問題なく成功すればバイナリファイルの「minimize.tpr」が生成されます。
gmx grompp -c initial.gro -p benzene_antechamber_GMX.top -o minimize -f minimize.mdpでは、実際に計算を実行しましょう!実行は以下のコマンドでできます。「-nt 2 -ntmpi 1 -ntomp 2」の部分のみ先に解説します。「-nt 2」は並列数を示していて、並列数を大きくすれば基本的には計算が早く終わります。可能な並列数はお使いのPCのCPU数に依存します。このブログを参考にしてVirtualBoxに環境を作成したのであれば、「VirtualBoxへのAlmaLinuxインストール」で指定したプロセッサー数が最大並列数になります。これ以上の値にすると計算が遅くなってしまうので注意してください。
「-ntmpi 1 -ntomp 2」はそれぞれMPIスレッド数とOpenMPスレッド数を指定しています。重要なのは、「MPIスレッド数×OpenMP=並列数」になるように設定が必要です。ここではMPIスレッド数を1、OpenMP=並列数としています。計算を効率よく実行するには適切な設定が必要ですが、まずはMPIスレッド数を1かつOpenMP=並列数にしておけば問題ないです。
gmx mdrun -nt 2 -ntmpi 1 -ntomp 2 -s minimize.tpr -deffnm minimize計算が完了すると、以下の4つのファイルが生成されているはずです。次のNVT計算に必要なのは、minimize.groになります。このファイルには、エネルギー極小化計算の最終的な構造データが格納されており、次の計算ではこの構造からスタートさせていきます。計算の詳細を確認したい場合は、minimize.logから確認できます。
・minimize.edr
・minimize.log
・minimize.gro
・minimize.trr
次に、NVT計算を行います。テキストファイルを「NVT.mdp」の名前で作成し、以下の内容をコピペしてください。
;##### Run Control ###############
integrator=md
dt=0.001
nsteps=100000
;##### Output control ############
nstxout=1000
nstvout=1000
nstlog=1000
nstcalcenergy=1000
nstenergy=1000
nstxtcout=0
energygrps=System
;##### Neighbor searching ########
pbc=xyz
rlist=1.0
verlet-buffer-tolerance=0.005
cutoff-scheme=Verlet
nstlist=10
ns-type=grid
;##### Van der Waals #############
vdwtype=cut-off
vdw-modifier=Potential-shift-Verlet
rvdw=1.0
DispCorr=Enerpres
;##### Electrostatics ############
coulombtype=PME
coulomb-modifier=Potential-shift-Verlet
rcoulomb=1.0
;##### Ewald #####################
fourierspacing=0.1
pme_order=4
ewald-rtol=1e-06
;##### Temperature coupling ######
tcoupl=Nose-hoover
nsttcouple=1
nh-chain-length=1
print-nose-hoover-chain-variables=yes
tc-grps=System
tau-t=0.6
ref-t=300
;##### Pressure coupling #########
pcoupl=no
;##### Simulated annealing #######
annealing=no
;##### Velocity generation #######
gen-vel=yes
gen-temp=300
gen-seed=-1
;##### Bonds #####################
continuation=no
constraints=none
;#################################エネルギー極小化計算の場合と同様に、.tprファイルを以下コマンドで作成します。
gmx grompp -c minimize.gro -p benzene_antechamber_GMX.top -o NVT -f NVT.mdp.tprファイルが生成できたら、同様に計算を以下コマンドで実行します。
gmx mdrun -nt 2 -ntmpi 1 -ntomp 2 -s NVT.tpr -deffnm NVT次に、NPT計算を行います。テキストファイルを「NPT.mdp」の名前で作成し、以下の内容をコピペしてください。
;##### Run Control ###############
integrator=md
dt=0.001
nsteps=100000
;##### Output control ############
nstxout=1000
nstvout=1000
nstlog=1000
nstcalcenergy=1000
nstenergy=1000
nstxtcout=0
energygrps=System
;##### Neighbor searching ########
pbc=xyz
rlist=1.0
verlet-buffer-tolerance=0.005
cutoff-scheme=Verlet
nstlist=10
ns-type=grid
;##### Van der Waals #############
vdwtype=cut-off
vdw-modifier=Potential-shift-Verlet
rvdw=1.0
DispCorr=Enerpres
;##### Electrostatics ############
coulombtype=PME
coulomb-modifier=Potential-shift-Verlet
rcoulomb=1.0
;##### Ewald #####################
fourierspacing=0.1
pme_order=4
ewald-rtol=1e-06
;##### Temperature coupling ######
tcoupl=Nose-hoover
nsttcouple=1
nh-chain-length=1
print-nose-hoover-chain-variables=yes
tc-grps=System
tau-t=0.6
ref-t=300
;##### Pressure coupling #########
pcoupl=Parrinello-Rahman
pcoupltype=isotropic
tau-p=2.0
ref-p=1.0
compressibility=4.5e-05
nstpcouple=1
;##### Simulated annealing #######
annealing=no
;##### Velocity generation #######
gen-vel=no
gen-temp=300
gen-seed=-1
;##### Bonds #####################
continuation=no
constraints=none
;#################################NVT計算の場合と同様に、.tprファイルを以下コマンドで作成します。
gmx grompp -c NVT.gro -p benzene_antechamber_GMX.top -o NPT -f NPT.mdp.tprファイルが生成できたら、同様に計算を以下コマンドで実行します。
gmx mdrun -nt 2 -ntmpi 1 -ntomp 2 -s NPT.tpr -deffnm NPTここまでが緩和計算になります。分子の集合体がある程度、安定な状態になってきています。
平衡化計算
次に平衡化計算を行います。NPT.mdpをeq.mdpの名前でコピーして、nsteps=1000000へ変更してください。その後の手順はNPT計算の場合と同様で、.tprファイルの作成、計算の実行と進めてください。NPT計算より計算時間が長くなっているので、終わるまでそこそこ時間がかかるかと思います。
gmx grompp -c NPT.gro -p benzene_antechamber_GMX.top -o eq -f eq.mdp
gmx mdrun -nt 2 -ntmpi 1 -ntomp 2 -s eq.tpr -deffnm eq計算が完了したら、平衡化しているかを確認しましょう。.edrファイルには計算の各ステップにおけるエネルギーや密度といった情報が格納されています。.edrファイルはバイナリファイルなので、このままでは中を見ることはできません。以下コマンドを実行してください。
gmx energy -f eq.edrすると、以下のようなメッセージが表示されます。それぞれに対応した番号を入力することで、計算結果を出力することができます。ここでは、「11 Potential」と「22 Density」を見てみましょう。「11」と入力してEnter、「22」と入力してEnter、最後に「0」を入力するとenergy.xvgが出力されます。
Select the terms you want from the following list by
selecting either (part of) the name or the number or a combination.
End your selection with an empty line or a zero.
-------------------------------------------------------------------
1 Bond 2 Angle 3 Proper-Dih. 4 Per.-Imp.-Dih.
5 LJ-14 6 Coulomb-14 7 LJ-(SR) 8 Disper.-corr.
9 Coulomb-(SR) 10 Coul.-recip. 11 Potential 12 Kinetic-En.
13 Total-Energy 14 Conserved-En. 15 Temperature 16 Pres.-DC
17 Pressure 18 Box-X 19 Box-Y 20 Box-Z
21 Volume 22 Density 23 pV 24 Enthalpy
25 Vir-XX 26 Vir-XY 27 Vir-XZ 28 Vir-YX
29 Vir-YY 30 Vir-YZ 31 Vir-ZX 32 Vir-ZY
33 Vir-ZZ 34 Pres-XX 35 Pres-XY 36 Pres-XZ
37 Pres-YX 38 Pres-YY 39 Pres-YZ 40 Pres-ZX
41 Pres-ZY 42 Pres-ZZ 43 #Surf*SurfTen 44 Box-Vel-XX
45 Box-Vel-YY 46 Box-Vel-ZZ 47 T-System 48 Xi-System
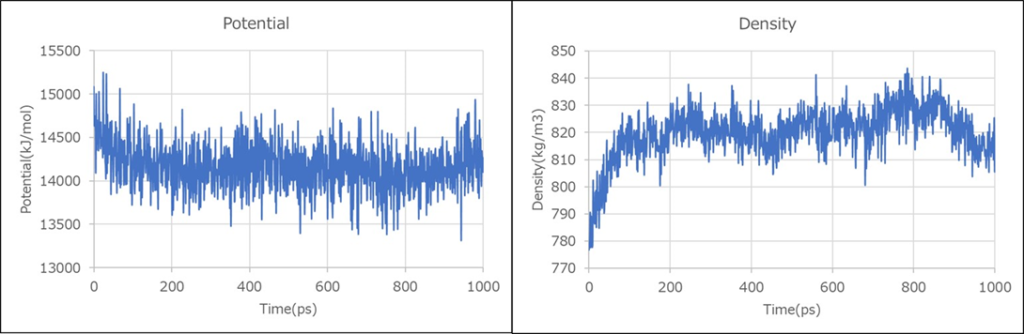
49 vXi-Systemenergy.xvgの中身を見てみましょう。以下のような部分があるかと思います。「0.000000 15083.195312 776.773438」の部分は、左から時間、ポテンシャルエネルギー、密度となっています。1000psの計算を実行して、1ps間隔でデータが出力されています。
@ xaxis label "Time (ps)"
@ yaxis label "(kJ/mol), (kg/m^3)"
0.000000 15083.195312 776.773438
1.000000 14643.376953 778.511230
2.000000 14748.033203 782.878235
3.000000 14626.832031 790.577026
・
・
・
998.000000 14263.921875 825.208679
999.000000 14100.155273 805.544922
1000.000000 14262.939453 818.976318時間に対して、ポテンシャルエネルギーと密度をプロットしてみましょう。計算の途中から、同じ値の前後で揺らいでいるのがわかるかと思います。密度はスタート時は780でしたが、途中から820前後を揺らいでいます。平衡状態に到達したかどうかの判断基準として、一定の範囲内で値が収まっているかどうかを見ることが重要になります。
ポテンシャルエネルギーと密度の値が大体同じ範囲で収まっているので、今回はこれで平衡状態に到達したとして進めます。ただし、実際に様々な物性を予測していくにあたっては、もっと平衡化計算に時間をかけて、平衡状態に到達したかどうかの判断を厳しめにすることを推奨します。平衡化の判断は物性計算にあたって非常に重要なので、その解説記事も今後書いていきます。
サンプリング計算
最後に、サンプリング計算に進みます。eq.mdpをsampling.mdpの名前でコピーして、平衡化計算の場合と同様に.tprファイルの作成、計算の実行と進めてください。
gmx grompp -c eq.gro -p benzene_antechamber_GMX.top -o sampling -f sampling.mdp
gmx mdrun -nt 2 -ntmpi 1 -ntomp 2 -s sampling.tpr -deffnm sampling計算が完了したら、「gmx energy -f sampling.edr」で密度がどのくらいになったか見てみましょう。密度を指定して実行すると、以下のような内容が表示されると思います。各ステップの密度の平均値が819.755(kg/m3)となっています。
Energy Average Err.Est. RMSD Tot-Drift
-------------------------------------------------------------------------------
Density 819.755 1.6 7.24992 6.62666 (kg/m^3)実際のベンゼンの密度は876kg/m3なので、やや実験値から外れていますね。実験値とずれている原因や対処方法については、解説記事にまとめていきます。
おわりに
今回は、前回の記事「GROMACS計算の下準備 ~分子構造作成・力場割り当て・分子の集合体の準備~」で作成したインプットを使って、GROMACSによる分子動力学計算を実行しました。分子動力学計算の流れが何となく掴めたであれば幸いです。
次回からは、前回の記事「GROMACS計算の下準備 ~分子構造作成・力場割り当て・分子の集合体の準備~」と、今回の記事に関する解説を書いていく予定です。


コメント
とても勉強になる記事をありがとうございます。質問なのですが、gromacsでの分子動力学計算のプログラム(ソースコード)を見ることは出来るのでしょうか。もしやり方をご存知でしたら、方法や参考になるページ等を教えていただけますと幸いです。
ご質問ありがとうございます。ソースコードはgithub(https://github.com/gromacs/gromacs)にアップされているので、そちらから確認できます。私はGROMACSのソースコードをあまり読んだことはないですが、MDで使うメインのコードの大半はsrc/gromacs/に入っているはずです。ソースコードのアルゴリズムについては、公式のマニュアルを参照頂くのが良いかと思います(https://manual.gromacs.org/2024.5/reference-manual/algorithms/algorithms.html#)。
お返事が遅れて申し訳ありません。教えていただきありがとうございました。次回の記事も楽しみにしております。
こんにちは。質問なのですが、gromacsのMDのプログラムを自分で書き換えて実行することはできるのでしょうか。(例えば解析に使いたいデータを出力するためにそれを計算するコードを数行書き足し、それを出力させる、など。)
ご質問ありがとうございます。はい、自分でプログラムを書き換えて実行可能です。ソースコードはgithub(https://github.com/gromacs/gromacs)にアップされていますので、必要な部分を書き足してインストールすればOKです。インストールの仕方は過去記事を参照頂ければと思います。