
今回は、実際にGROMACSで分子動力学計算を行うための下準備を行っていきます。以前の記事「分子動力学法の計算の流れ」で計算の流れを説明しました。この流れに沿って、下準備のやり方を書いていきます。分子動力学法の下準備は手間がかかり、ややこしい部分も多々ありますが、まずはこの記事に沿って試してみてください。一つ一つの流れを詳しく理解するのは後からでも大丈夫ですので、一旦最後までやってみると分子動力学法に対する理解がしやすくなると思います。基本的には過去記事を参考に環境を作った前提で進めますので、わからない場合は過去記事を参照してください。
計算したい内容を決める
では、まず計算したい内容を決めましょう!いきなり複雑な現象を計算するのは大変なので、今回は簡単な課題を設定します。『液体のベンゼンの密度を計算によって求める』、これを目的に進めていきますね。ベンゼンの密度なんて調べればすぐわかるじゃん!って言われそうですが、その通りです。ただ、言い換えればすぐに実験データが見つかるので、計算結果が実験を再現できているかが容易に確認できますよね。ということで、この課題で進めていきましょう。
分子構造の作成
最初に、分子構造を作成します。以前の記事でも書きましたが、「液体のベンゼンの密度を計算したい」のであれば、ベンゼンの分子構造の情報が入った「何か」を準備する必要があります。今回はmolファイルといった形式で準備していきましょう。molファイルの準備の仕方は多岐にわたりますが、比較的容易に実施しやすいMolViewで作る方法を紹介します。
MolViewはブラウザ上で利用できる、分子構造を描画できるアプリです。まずはMolViewにアクセスしてみましょう、リンクを貼っておきます。私はGoogle Chromeでアクセスしています、他のブラウザで上手くいかなかったら変えてみてください。
MolViewリンク:https://molview.org/
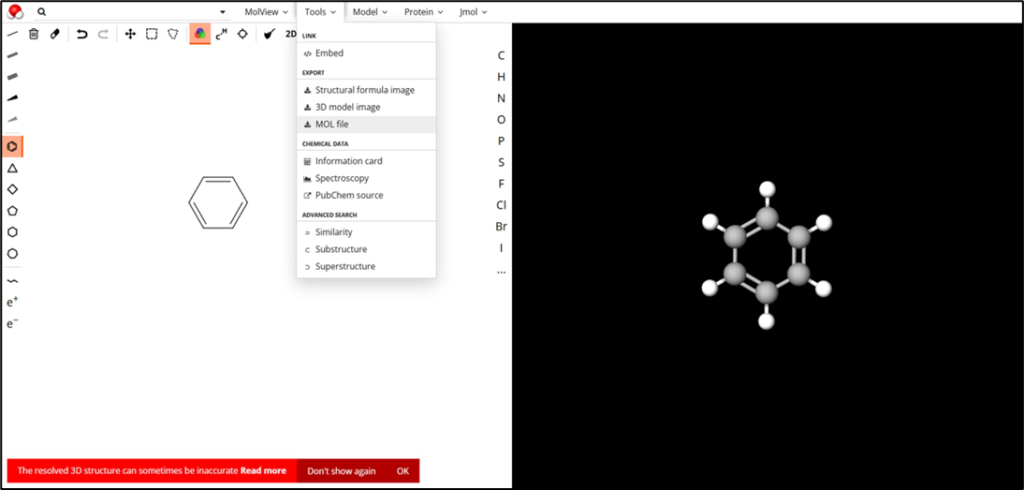
アクセスすると、下図のように既に分子構造が描画された画面が表示されます。
一旦この分子構造を削除するため、左上のゴミ箱アイコン(Clear all)をクリックして、左のキャンパス画面をきれいにしましょう。
次に、ベンゼン環のアイコンをクリックして、キャンパス画面をクリックします。ベンゼン環のアイコンは下図の一番左の部分にあります。
キャンパス画面にベンゼン環が表示されたら、次に「2D to 3D」をクリックして、右側の分子構造を表示する画面で作成した構造を確認します。下図のようになっていればOKです。
最後に、作成した分子構造をmolファイルとして取得しましょう。「Tools」の「MOL file」を選択して、molファイルをダウンロードします。わかりやすいように、ファイル名をbenzene.molに修正しておきます。これで、分子構造の準備は完了です!
力場の割り当て
次に、力場の割り当てをします。これは、以前の記事で紹介したacpypeを使っていきます。acpypeのインストール方法は過去記事を参照してください。
まず、先ほど作ったbenzene.molをacpypeがインストールされている環境に持っていってください。ここまでの記事を参考に進めていたのであれば、VirtualBoxの仮想環境にWinSCPで持っていけばOKです。ファイルがたくさん生成されるので、作業用のフォルダを作成しておくことを推奨します。
次に、acpypeがインストールされたPython仮想環境をアクティベートしましょう。以前の記事では「SimulationEnv」という名前でPython仮想環境を構築しましたので、これをアクティベートします。アクティベートは以下のコマンドで実施してください。
conda activate SimulationEnv次に、molファイルをmol2ファイルに変換します。mol2ファイルもmolファイルと同様に分子構造のデータを含むファイルです。この後の工程ではmol2ファイルを求められるので、ここで変換します。変換の仕方は以下のコマンドを実行すればOKです。openbabelというプログラムを使って変換しています、benzene.molがインプット、benzene.mol2がアウトプットの部分になります。benzene.mol2が生成されていればOKです。ちなみに、Tera Term上で実行していると思いますが、「ls」と入力すれば現在ディレクトリのファイル一覧が表示されるので便利です。
obabel benzene.mol -O benzene.mol2次に、mol2ファイルを力場が割り当てられるよう原子タイプを設定します。これにはantechamberというプログラムを使います。acpypeをインストールしていれば自動的にインストールされています。原子タイプの設定は以下のコマンドを実行すればOKです。benzene.mol2がインプット、benzene_antechamber.mol2がアウトプットの部分になります。それ以外の内容は、別途解説記事を書いていく予定なので、一旦は次に進みましょう。
antechamber -i benzene.mol2 -fi mol2 -o benzene_antechamber.mol2 -fo mol2 -c gas -nc 0 -m 1 -at gaff最後に、力場を割り当てたファイルを生成します。acpypeを使って、先ほどantechamberで生成したmol2ファイルから作成します。作成の仕方は以下のコマンドを実行すればOKです。benzene_antechamber.mol2がインプットです。antechamber同様、他の内容は別途解説記事を書いていく予定です。コマンドを実行すれば、「benzene_antechamber.acpype」というフォルダが生成しているはずです。
acpype -i benzene_antechamber.mol2 -a gaff -c gas フォルダ「benzene_antechamber.acpype」の中にはたくさんのファイルが生成されますが、その中で今回の計算に必要となるのが以下の3つです。これらを作業をしていたフォルダにコピーしてください。
・benzene_antechamber_GMX.top
・benzene_antechamber_GMX.gro
・benzene_antechamber_GMX.itp
benzene_antechamber_GMX.topを少し修正するので、このファイルを開いてください。まず、以下の部分は不要なので削除してください。
; Ligand position restraints
#ifdef POSRES_LIG
#include "posre_benzene_antechamber.itp"
#endifさらに、以下のようにnmolsのところに1が設定されています。これはベンゼンを1分子、シミュレーションを行うボックスに入れるということを示しています。この後の工程で、ベンゼン500分子を入れたボックスを作成するので、この値を500に変更してください。これで、力場の割り当ては完了です。
[ molecules ]
; Compound nmols
benzene_antechamber 1 分子の集合体の準備
次に、分子の集合体の準備をします。これにはpackmolというプログラムを使います、acpypeをインストールしていれば自動的にインストールされています。
まず、先ほど作成した「benzene_antechamber_GMX.gro」をpdb形式のファイルに変換します。これはGROMACSを使って変換でき、以下のコマンドを実行すればOKです。
gmx editconf -f benzene_antechamber_GMX.gro -o benzene_antechamber_GMX.pdb次に、packmolのインプットを作成します。「packmol.in」という名前でテキストファイルを生成し、以下の内容を入力してください。benzene_antechamber_GMX.pdbを500個、各辺が50Åの立方体に入れていくことを示しています。packmolについても、別途解説記事を書いていく予定です。
tolerance 2.0
filetype pdb
structure benzene_antechamber_GMX.pdb
number 500
inside box 0.0 0.0 0.0 50.0 50.0 50.0
end structure
output packmol_out.pdbpackmol.inを作成したら、以下コマンドを実行してください。無事に成功すれば、packmol_out.pdbが生成しているはずです。
packmol < packmol.in最後に、生成したpackmol_out.pdbをgroファイルに変換します。groファイルは、シミュレーションボックスに分子を入れた状態の構造データと理解しておけばここでは大丈夫です。変換の際、シミュレーションボックスのサイズをnm単位で指定します。GROMACSでは、長さを基本的にnm単位で表すので注意して下さい。
gmx editconf -f packmol_out.pdb -box 5.0 5.0 5.0 -o initial.groinitial.groが生成していれば、分子の集合体の準備は完了です。
おわりに
今回は分子動力学法の計算の下準備のやり方を紹介しました。計算の内容によって設定するオプションは変わってきますが、基本的な流れは同じケースが多いです。次回の記事と合わせて、下準備から計算の実行まで一連の流れを体験できれば、分子動力学法がどういったものかイメージできるようになるかと思います。オプションの内容や、それぞれの工程の意味はその後少しずつ理解していけば大丈夫です。最初から全て理解しようとするよりも、一旦全体感を把握してから個々を学んでいくのがおススメです。


コメント
非常に分かりやすくて勉強になります。
質問があるのですが、こちらの記事で紹介されている方法で割り当てられる力場はいわゆるAMBER力場(?)だと思うのですが、OPLS-UAやOPLS-AA、または剛体モデルを使いたい場合は、それぞれどのようにすればよいのでしょうか。
ご質問ありがとうございます。仰る通り、gaff(General Amber Force Field)はAMBER形式の力場です。私はOPLS力場を普段使っていないので、わかる範囲ですが回答いたします。
まず、上記手順の「acpype -i benzene_antechamber.mol2 -a gaff -c gas 」を実行した際、フォルダ「benzene_antechamber.acpype」が生成され、その中に「benzene_antechamber_GMX_OPLS.itp」があります。これはOPLS-AA力場を割り当てたitpファイルですが、これを使うのは推奨しません。「acpype -h」を実行すると、「root_GMX_OPLS.itp : OPLS/AA mol unit topol & par file for GROMACS (experimental!)」を記載があるように、動作保証がきちんとされていない模様です。LJポテンシャルのパラメータも自動設定されていないので、使いにくいです。
OPLS-AAを使うのであれば、簡単なのはLigParGenを使うのが良いと思います。サイトにアクセスして、molファイル等の分子構造をアップロード、電荷の設定をしてSubmitすればGROMACS用のitpを生成することができます。
LigParGen:https://zarbi.chem.yale.edu/ligpargen/
他の方法としては、moltemplateを使ってOPLS-AAを割り当てることもできます。ただ、こちらはLAMMPS用のフォーマットになるので、LAMMPS用からGROMACS用に変換する手間がかかります。そのため、まずはLigParGenを使うのが良いと思います。
OPLS-UAについては、すみませんがほとんど知見がないです。ちょっと調べてみたところ、パラメータ等で参考になりそうなリンクがあったので貼っておきます。自動で力場を割り当てるプログラムがない場合は、OPLS-AAで生成したitpを手動で変更する、もしくはPython等で自作するしかないです。
参考リンク:https://towhee.sourceforge.net/forcefields/oplsua.html
剛体モデルはSPCやTIP3Pのような一般的なものであれば、GROMACSのソースコードに同梱されています。「/share/top/amber99.ff/tip3p.itp」のように入っているので、必要な剛体モデルがあるかご確認してみてください。
ありがとうございます。また疑問点が見つかったら教えてください。
記事の更新、楽しみにしております。
コメント失礼いたします。複数の成分(例えばベンゼンとトルエンを250個ずつ混ぜた系など)でシミュレーションしたい場合はどのようにすれば良いのでしょうか。この次の記事の「GROMACS計算の実行」で行う手順は一成分系でも多成分系でも変わらないかと思っているのですが、本記事で解説されている段階でどのように行えばよいのか、教えて頂きたいです。
ご質問ありがとうございます。複数成分系の場合はtopファイル等を一部変更する必要があります。少し長文になってしまいますので、複数成分系の場合の変更点をまとめて記事にしました、そちらを参考にして頂ければと思います。
https://start-pc-simulation.com/archives/365