
今回は、分子動力学法による計算の流れについて書いていきます。シミュレーションができる環境が整っても、どのように計算を進めていけば良いのかわからないケースは多々あると思います。そのため、実際の計算を始める前に、一度全体の流れを把握することをお勧めします。今回紹介する流れはあくまで一般的な流れであるので、ケースによってはやり方を変える必要があります。ただ、一般的な流れを知っていないと特殊なケースに対応することもできないので、今回の記事の内容を知っておいて損はないと思います。
計算全体の流れ
一般的な分子動力学法による計算全体の流れを下図にまとめました、各プロセスを簡単に解説していきますね。青色のプロセスは分子動力学法を始める前の下準備、緑色のプロセスは分子動力学法を使った計算のプロセスです。
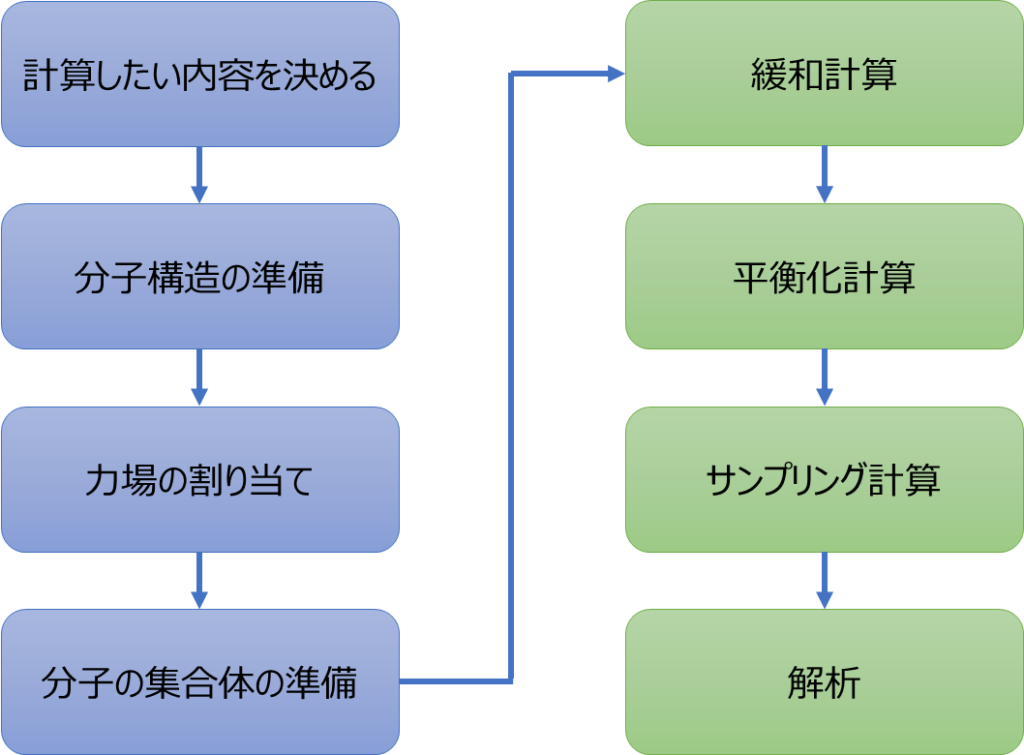
① 計算したい内容を決める
まず最初に、分子動力学法によって何を計算したいかを決めます。例えば、拡散係数や密度、誘電率といった物性ですね。物性によっては上図のような一般的な流れを少し変えないといけない場合がありますので、この流れで問題ないかを物性ごとに理解しておくことが必要です。ここでは、この後の説明が理解しやすいように「液体のベンゼンの密度を計算したい」として進めていきますね。
② 分子構造の準備
次に、分子構造の準備が必要です。「液体のベンゼンの密度を計算したい」のであれば、ベンゼンの分子構造の情報が入った「何か」を準備する必要があります。この「何か」ですが、良く使われる例を挙げるとSMILESやmolファイルといったものがあります。これ以降のプロセスに対応できる形式であれば何でも大丈夫です。準備の仕方は今後の記事で紹介しますので、ここではそういうものが必要なんだっていう理解をして頂ければOKです。
③ 力場の割り当て
分子構造の準備ができたら、それに力場を割り当てることが必要です。力場というのは、分子動力学法で分子内や分子間に働く力を計算するために必要なパラメータです。具体的な説明をすると長くなるので今回は割愛しますが、分子動力学法で必須となる非常に重要なパラメータということを、まずは覚えていてください。この力場の割り当て方法も今後の記事で紹介します。
④ 分子の集合体の準備
力場を割り当てたら、次に分子の集合体を準備します。一般的に分子の物性は1分子ではなく、大量の分子が集まった状態で得られるものになります。「液体のベンゼンの密度を計算したい」のであれば、大量のベンゼン分子が集まって液体を形成している状態を再現しなければならないので、それに近い状態を事前に準備してあげる必要があります。いきなり液体と同じ状態を再現することはできないので、あくまで近い状態を準備し、分子動力学法により実際の液体の状態にできるだけ近づけていくという流れになります。この分子の集合体の準備方法も今後の記事で紹介します。
⑤ 緩和計算
ここからが分子動力学法による計算、つまり前回記事まででインストールしたGROMACSを用いた計算になります。緩和計算というのは、分子を軽く動かして不安定な状態からある程度安定な状態に持っていく計算のことです。教科書や論文によっては、緩和計算と平衡化計算をまとめて緩和計算と呼んだり、平衡化計算を呼ぶこともありますが、ここでは分けて説明します。
事前に準備した分子の集合体は大抵の場合、分子が無理のある構造をしていたり、分子同士が近い、もしくは極端な場合は重なっていることが多々あります。このまま平衡化計算をしようとすると、分子の集合体が爆発します(笑)実際に爆発しているわけではないですが、挙動として爆発しているかのごとく、分子同士が離れて吹き飛んでいきます。このまま計算を進めていっても、分子同士が離れた状態のままであり、気体に近い状態になってしまいます。「液体のベンゼンの密度を計算したい」という目的から異なった計算になってしまいますね。
なぜこのようになるかというと、分子同士が接近しすぎて反発する大きな力が働いてしまい、一気に離れようとするからです。これを避けるために、分子を軽く動かして無理のない構造にしつつ、近すぎる分子は少し離してあげることをします。これが緩和計算の目的です。
⑥ 平衡化計算
緩和計算が終わったら平衡化計算に進みます。これは文字通り、平衡状態に持っていく計算となります。「液体のベンゼンの密度を計算したい」という場合、当然ながら知りたいのは平衡状態の密度ですよね。SDSとか様々なデータベースに記載されている密度は、各温度条件において平衡状態となった際の密度です。
設定した計算条件において計算を進めていくことで、どんどん分子の集合体が安定な状態に向かっていき、最終的には平衡状態となります。平衡状態になったかどうかはエネルギーであったり、密度がほとんど変動しなくなったことから判断することが多いです。この判断を誤ってしまうと、得られる物性が計算するたびに大きく変わったりしてしまい、間違った考察につながってしまう危険があります。そのため、平衡になったかどうかをきちんと判断することは非常に重要です。
⑦ サンプリング計算
平衡状態になったのを確認したら、サンプリング計算を進めます。サンプリング計算を簡単に説明すると、ある程度の長時間計算を行い、たくさんの状態を採取する計算となります。まず、分子の集合体はずっと同じ状態をしているわけではありません。現実世界で目の前の液体を見ると何も変化していないように見えますが、実際には分子は常に動き回っています。固体状態であったとしても分子は完全に静止しているわけではなく、振動して動いています。
絶えず動いているということにより、平衡状態であっても液体の密度は見る瞬間によって違った値となります。「じゃあどのタイミングの密度を使えば良いのか?」と思われるかもしれませんが、そもそも一つのタイミングで密度を決めることが間違いです。計算の話に戻りますが、ある時刻で密度を取得、少し計算を進めた点で再度取得、また計算を進めた点で再度取得、ということを延々と繰り返していきます。最終的に、大量の密度データが得られることになり、この平均値を計算の結果として用います。このようにすることで、計算するたびに物性が大きく変わることなく、安定して物性を得ることが可能となります。
⑧ 解析
サンプリング計算が完了したら、その結果を解析します。「液体のベンゼンの密度を計算したい」という内容であれば、そもそも密度は計算の結果として直接出力されるので解析は必要ないです。しかし、物性によっては計算の結果から得られないこともあるので、得られた計算の結果を解析して物性を得るというプロセスが必要になります。どのように解析するかは物性によって様々ですが、出力される分子の動きを用いて解析することが多いです。
解析するための機能が分子動力学法のプログラムに準備されている場合もありますが、ない場合は自分で解析プログラムを作る、もしくは公開されている解析プログラムを探して用いるなどをしなければいけません。自分で解析プログラムを作る方法であったり、公開されている解析プログラムの使い方は今後の記事で紹介していきます。
おわりに
今回は分子動力学法による計算の流れを紹介しました。あくまで一般的な流れではありますが、知っておいて損はない内容なので、参考にして頂ければ幸いです。次回はこの記事で紹介した青色のプロセス部分、分子動力学法を始める前の下準備についてやり方を書いていきます。


コメント